






{kind=link}
{kind=link}
{kind=link}
{kind=link}

If you find GSearch useful, please cite the following paper:
@article{zhao2024gsearch,
title={GSearch: ultra-fast and scalable genome search by combining K-mer hashing with hierarchical navigable small world graphs},
author={Zhao, Jianshu and Both, Jean Pierre and Rodriguez-R, Luis M and Konstantinidis, Konstantinos T},
journal={Nucleic Acids Research},
volume={52},
number={16},
pages={e74--e74},
year={2024},
publisher={Oxford University Press}
}
conda install -c conda-forge -c bioconda gsearch=0.2.2wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.5/gsearch_Linux_x86-64_v0.1.5.zip
unzip gsearch_Linux_x86-64_v0.1.5.zip
chmod a+x ./gsearch_Linux_x86-64_v0.1.5/*
cd gsearch_Linux_x86-64_v0.1.5
./gsearch -hNote that pre-built databases will not work for development version, you need to rebuild database yourself
conda install -c bioconda -c conda-forge gsearch
##or via cargo, install cargo:
curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh
## install via cargo
cargo install gsearch --features annembed_intel-mkl,simdeez_f --force
## For macOS see below install section for details
GSearch stands for genomic search.
This package (currently in development) compute MinHash-like signatures of bacteria and archaea (or virus and fungi) genomes and stores the id of bacteria and MinHash-like signatures in a Hnsw structure for searching of new request genomes. A total of ~50,000 to ~60,000 lines of highly optimized Rust code were provided in this repo and several other crates/libraries develped for this repo, such as kmerutils, probminhash, hnswlib-rs and annembed, see below for details. Some of the libraries are very popular and have been used about ~40 thousand times, see here.
This package is developped by Jean-Pierre Both jpboth for the software part and Jianshu Zhao for the genomics part. We also created a mirror of this repo on GitLab and Gitee (You need to log in first to see the content), just in case Github service is not available in some region.
gsearch -h
************** initializing logger *****************
Approximate nearest neighbour search for microbial genomes based on MinHash-like metric
Usage: gsearch [OPTIONS] [COMMAND]
Commands:
tohnsw Build HNSW graph database from a collection of database genomes based on MinHash-like metric
add Add new genome files to a pre-built HNSW graph database
request Request nearest neighbors of query genomes against a pre-built HNSW graph database/index
ann Approximate Nearest Neighbor Embedding using UMAP-like algorithm
help Print this message or the help of the given subcommand(s)
Options:
--pio <pio> Parallel IO processing
--nbthreads <nbthreads> nb thread for sketching
-h, --help Print help
-V, --version Print version
The objective is to use the Jaccard index as an accurate proxy of mutation rate or Average Nucleitide Identity(ANI) or Average Amino Acide Identity (AAI) According to equation (Poisson model or Binomial model):
or
where J is Jaccard-like index (e.g. Jp from ProbMinHash or J from SuperMinHash, SetSketch or Densified MinHash, a class of locality sensitive hashing algorithms, suitable for nearest neighbor search, see below) and k is k-mer size. To achieve this we use sketching. We generate kmers along genome DNA or amino acid sequences and sketch the kmer distribution encountered (weighted or not) in a file, see kmerutils. Then final sketch is stored in a Hnsw database See here hnsw or here hnsw.
The sketching and HNSW graph database building is done by the subcommand tohnsw.
The Jaccard index come in 2 flavours:
1. The probability Jaccard index that takes into account the Kmer multiplicity. It is defined by :
The above mentioned choices for sketching can be specified in "gsearch tohnsw" subcommand via the --algo option (prob, super/super2, hll and optdens). We suggest using either ProbMinHash or Optimal Densification. For SuperMinhash, 2 implementations are available: super is for floating point type sketch while super2 is for integer type sketch. The later is much faster than the former. hll is for SetSketch, we name it hll because the SetSketch data structure can be seen as a similar implementation of HyperLogLog, but adding new algorithms for similarity estimation (e.g., Locality Sensitivity, LSH or Joint Maximum Likelihood Estimation, JMLE for Jaccard index) in additional to distinct element counting. Also becasuse we think HyperLogLog is a beautiful name, which describes the key steps of the implementation.
The estimated Jaccard-like index is used to build HNSW graph database, which is implemented in crate hnswlib-rs.
The sketching of reference genomes and HNSW graph database building can take some time (less than 0.5 hours for ~65,000 bacterial genomes of GTDB for parameters giving a correct quality of sketching, or 2 to 3 hours for entire NCBI/RefSeq prokaryotic genomes. ~318K). Result is stored in 2 structures:
- A Hnsw structure storing rank of data processed and corresponding sketches.
- A Dictionary associating each rank to a fasta id and fasta filename.
The Hnsw structure is dumped in hnswdump.hnsw.graph and hnswdump.hnsw.data The Dictionary is dumped in a json file seqdict.json
************** initializing logger *****************
Build HNSW graph database from a collection of database genomes based on MinHash-like metric
Usage: gsearch tohnsw [OPTIONS] --dir <hnsw_dir> --kmer <kmer_size> --sketch <sketch_size> --nbng <neighbours> --algo <sketch_algo>
Options:
-d, --dir <hnsw_dir> directory for storing database genomes
-k, --kmer <kmer_size> k-mer size to use
-s, --sketch <sketch_size> sketch size of minhash to use
-n, --nbng <neighbours> Maximum allowed number of neighbors (M) in HNSW
--ef <ef> ef_construct in HNSW
--scale_modify_f <scale_modify> scale modification factor in HNSW or HubNSW, must be in [0.2,1] [default: 1.0]
--algo <sketch_algo> specifiy the algorithm to use for sketching: prob, super/super2, hll or optdens/revoptdens
--aa --aa Specificy amino acid processing, require no value
--block --block : sketching is done concatenating sequences
-h, --help Print help
For adding new genomes to existing database, the add subcommand is being used. It will automatically load sketching files, graph files and also paremeters used for building the graph and then use the same parameters to add new genomes to exisiting database genomes.
************** initializing logger *****************
Add new genome files to a pre-built HNSW graph database
Usage: gsearch add --hnsw <hnsw_dir> --new <newdata_dir>
Options:
-b, --hnsw <hnsw_dir> set the name of directory containing already constructed hnsw data
-n, --new <newdata_dir> set directory containing new data
-h, --help Print helpFor requests the subcommand request is being used. It reloads the dumped files, hnsw and seqdict related takes a list of fasta files containing requests and for each fasta file dumps the asked number of nearest neighbours according to distance mentioned above.
************** initializing logger *****************
Request nearest neighbors of query genomes against a pre-built HNSW graph database/index
Usage: gsearch request --hnsw <DATADIR> --nbanswers <nb_answers> --query <request_dir>
Options:
-b, --hnsw <DATADIR> directory contains pre-built database files
-n, --nbanswers <nb_answers> Sets the number of neighbors for the query
-r, --query <request_dir> Sets the directory of request genomes
-h, --help Print help
A tabular file will be saved to disk in current directory (gsearch.neighbors.txt) with 3 key columns: query genome path, database genome path (ranked by distance) and distance. The distance can be transformed into ANI or AAI according to the equation above. We provide the program reformat (also parallel implementation) to do that:
reformat -h
Processes input files for ANI calculation
Usage: reformat <kmer> <model> <input_file> <output_file>
Arguments:
<kmer> The kmer value used for ANI calculation (16)
<model> The model to be used for ANI calculation (1 or 2,corresponding to Poisson model and Binomial model)
<input_file> File containing the data to be transformed into tabular format
<output_file> File where the tabular output will be saved
Options:
-h, --help Print help
-V, --version Print versionreformat 16 1 ./gsearch.neighbors.txt ./clean.txtYou will then see the clean output like this:
| Query_Name | Distance | Neighbor_Fasta_name | Neighbor_Seq_Len | ANI |
|---|---|---|---|---|
| test03.fasta.gz | 5.40E-01 | GCF_024448335.1_genomic.fna.gz | 4379993 | 97.1126 |
| test03.fasta.gz | 8.22E-01 | GCF_000219605.1_genomic.fna.gz | 4547930 | 92.5276 |
| test03.fasta.gz | 8.71E-01 | GCF_021432085.1_genomic.fna.gz | 4775870 | 90.7837 |
| test03.fasta.gz | 8.76E-01 | GCF_003935375.1_genomic.fna.gz | 4657537 | 90.5424 |
| test03.fasta.gz | 8.78E-01 | GCF_000341615.1_genomic.fna.gz | 4674664 | 90.4745 |
| test03.fasta.gz | 8.79E-01 | GCF_014764705.1_genomic.fna.gz | 4861582 | 90.4108 |
| test03.fasta.gz | 8.79E-01 | GCF_000935215.1_genomic.fna.gz | 4878963 | 90.398 |
| test03.fasta.gz | 8.82E-01 | GCF_002929225.1_genomic.fna.gz | 4898053 | 90.2678 |
| test03.fasta.gz | 8.83E-01 | GCA_007713455.1_genomic.fna.gz | 4711826 | 90.2361 |
| test03.fasta.gz | 8.86E-01 | GCF_003696315.1_genomic.fna.gz | 4321164 | 90.1098 |
Query_Name is your query genomes, Distance is genomic Jaccard distance (1-J/Jp), Neighbor_Fasta_name is the nearest neighbors based on the genomic Jaccard distance, ranked by distance. ANI is calculated from genomic Jaccard distance according the equaiton above between you query genome and nearest database genomes. You can then simply extract the best one match according to the highest ANI from the above output:
awk 'BEGIN{FS=OFS="\t"} NR == 1 {print; next} {if (!max[$1] || $5 > max[$1]) {max[$1] = $5; line[$1] = $0}} END {for (x in line) print line[x]}' clean.txt > clean_best_hit.txtWe also provide scripts for analyzing output from request and compare with other ANI based methods here: https://github.com/jianshu93/gsearch_analysis
Extract nearest neighbor genomes in the pre-built database. For each genome, nearest neighbor genomes to it will printed from samllest distance (largest ANI) to largest distance (smallest ANI)
$hnsw2knn -h
************** initializing logger *****************
Extract K Nearest Neighbors (K-NN) from HNSW graph.
Usage: hnsw2knn [OPTIONS] --hnsw <DATADIR> --output <OUTPUT_PATH>
Options:
-b, --hnsw <DATADIR> Directory containing pre-built HNSW database files
-o, --output <OUTPUT_PATH> Output path to write the neighbor list
-n, --k-nearest-neighbors <KNN> Number of k-nearest-neighbors to extract [default: 32]
-h, --help Print help
-V, --version Print versionAdditional ANI calculation (if you do not want to use MinHash estimated ANI) for the query genomes and nearest neighbor genomes can be performed via the program superani:
superani -h
************** initializing logger *****************
Computing average nucleotide identity between reference and query genomes via sparse kmer chaining or Open Syncmer with Densified MinHash
Usage: superani --ql <FILE> --rl <FILE> --output <FILE>
Options:
-q, --ql <FILE> A file containing a list of query genome paths (.gz supported)
-r, --rl <FILE> A file containing a list of reference genome paths (.gz supported)
-o, --output <FILE> Output file to write results
-h, --help Print help
-V, --version Print versionThe input is query genome path and reference genome path and output is ANI between each query and each reference genome. Both input files can be created by extracting from the output of gsearch request command with some modification of genome path. Multithreaded computation is supported and it can also be used as a seperate ANI calculator.
### predict genes for request at amino acid level, credit to original author but we rewrite the user inteface to be consistent with gsearch and other commands
FragGeneScanRs -s ./data/NC_000913.fna -o NC_000913 -t complete -p 8
hmmsearch_rs -h
************** initializing logger *****************
Search protein sequences against HMM profiles
Usage: hmmsearch_rs [OPTIONS] --faa <fasta> --hmm <hmm>
Options:
-f, --faa <fasta> Path to the faa file containing the protein sequences
-m, --hmm <hmm> Path to the HMM file
-o, --output <output> Output file to save the search results
-h, --help Print help
-V, --version Print versionwe wrapped the HMMER C API and made modification so that the output is tabular for easy parsing. Universal gene HMMs can be found in the data folder. This command is only available in the release page for linux, not via bioconda. It is available for MacOS
### search a proteome agains a HMM pforile
hmmsearch_rs -f ./data/test03.faa -m ./data/HMM_bacteria/PF00380.20.HMMCalculate AAI between genomes based on FracMinhash
************** initializing logger *****************
Compute Average Amino Acid Identity (AAI) via FracMinHash/Sourmash for genomes
Usage: superaai [OPTIONS] --ql <FILE> --rl <FILE> --output <FILE>
Options:
-q, --ql <FILE> File containing list of query protein paths (.faa format, .gz supported)
-r, --rl <FILE> File containing list of reference protein paths (.faa format, .gz supported)
-o, --output <FILE> Output file to write results
-k, --kmer <INT> K-mer size for MinHash calculation [default: 7]
-l, --scaled <INT> Scaled factor for MinHash calculation [default: 100]
-s, --sketch <INT> Sketch size for MinHash (number of hashes) [default: 5120]
-h, --help Print help
-V, --version Print versionThe input is query genome path (proteome) and reference genome path (proteome) and output is AAI between each query and each reference genome.
For UMAP-like algorithm to perform dimension reduction and then visuzlizing genome database, we run it after the tohnsw step (pre-built database) (see below useage ann section). See annembed crate for details. Then the output of this step can be visualized, for example for the GTDB v207 we have the following plot. See paper here.
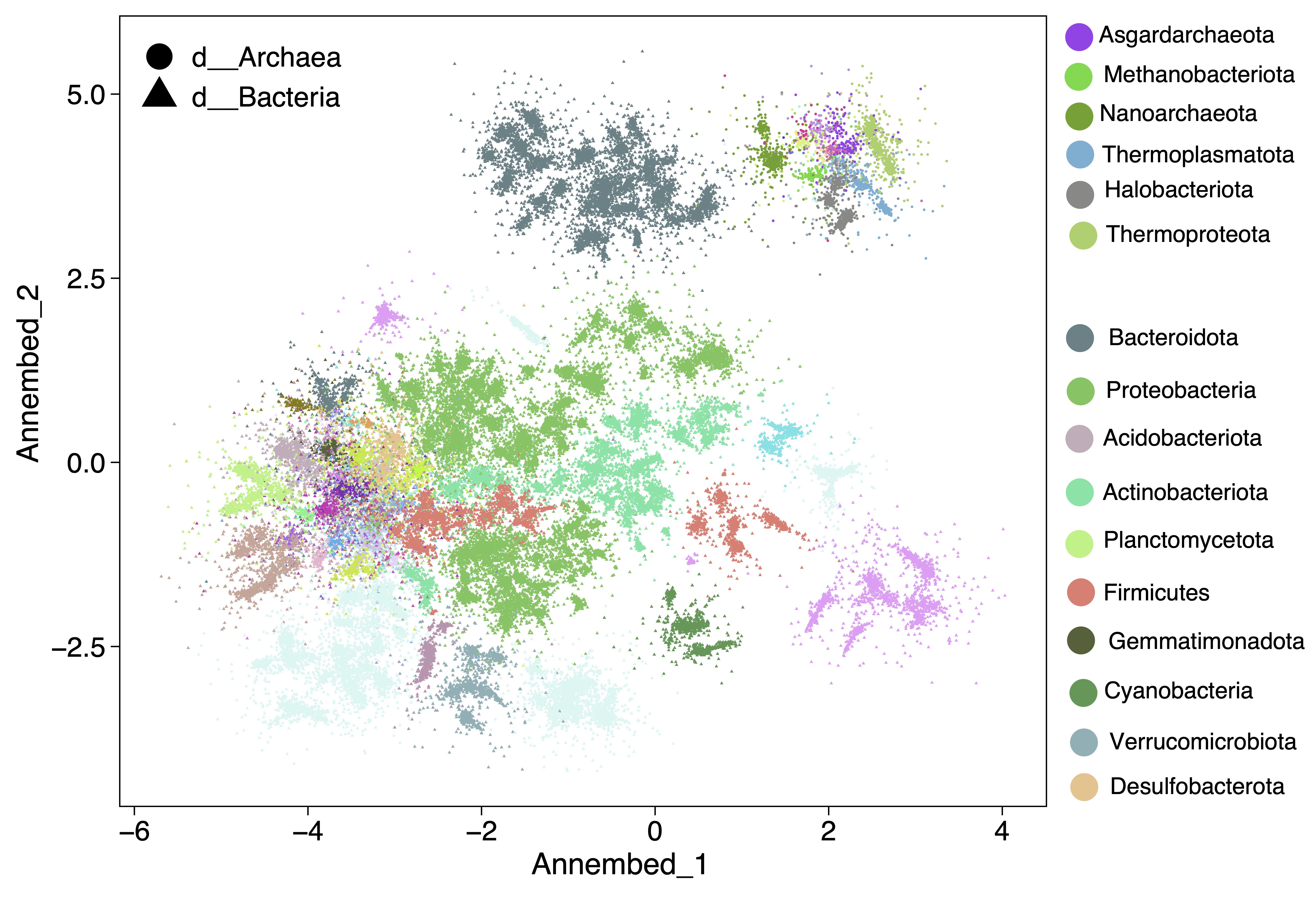
We provide a bunch of scripts to allow split database genomes into N pieces and build HNSW graph database for each piece, then run search of new genomes against those pieces and collect results. This will only requires 1/N memory for your machine at the cost of additional sketching for the same query genomes.
### for a folder with genomes in it, we can split it into N subfolders randomly by running:
./scripts/split_folder.sh input_folder_path output_folder_path 3
### using the output from above split_folder.sh step, build database for each subfolder
./scripts/multiple_build.sh output_folder_path gsearch_db_folder
### using output from above multiple_build.sh step, search new genomes againt each database
./scripts/multiple_search.sh gsearch_db_folder_output new_genome_folder_path outputFor real world datasets with billions of genomes, in additional to the random split idea mentioned above, approximate clustering via CoreSet can be used via the hnswcore command:
### for linux, you will also need to download the pre-compiled binary, for macOS brew install will be ok
wget https://github.com/jianshu93/coreset/releases/download/v0.1.0/hnswcore_Linux_x86-64_v0.1.0.zip
unzip hnswcore_Linux_x86-64_v0.1.0.zip
chmod a+x ./hnswcore- First, we need to build a database via MinHash sketch and have nearest neighbors for each database genome.
gsearch --pio 4000 --nbthreads 60 tohnsw -d ../files -k 16 -s 18000 -n 128 --ef 1600 --algo optdens
- Second, we use the output from the step 1 to run Coreset clustering, --dir is the current directory in step 1.
hnswcore -h
************** initializing logger *****************
Approximate Clustering via Streaming CoreSet Construction
Usage: hnswcore --dir <dir> --fname <fname> --type <typename> [COMMAND]
Commands:
coreset CoreSet Construction
help Print this message or the help of the given subcommand(s)
Options:
-d, --dir <dir> directory contains HNSW database files
-f, --fname <fname> HNSW database file basename
-t, --type <typename> type for HNSW distance, e.f., f32, u32
-h, --help Print helpYou can ask for a given number of cluters so that each cluster of genome sketches is smaller in size:
### if the optdens and revoptdens was used (see step 1), the output HNSW database can be clustered into 5 pieces like this
hnswcore --dir ./ --fname hnswdump --type f32 coreset --cluster 5- Use the dictjsontocsv.ipynb python notebook in scripts folder to transfrom the cluster membership information id in HNSW to actual genome id.
We also provide general purpose sequence search via BigSig(BItsliced Genomic Signature Index), which can be used to quickly identify reads against a reference genome database.
************** initializing logger *****************
Large-scale Sequence Search with BItsliced Genomic Signature Index (BIGSIG)
Usage: bigsig [COMMAND]
Commands:
construct Construct a BIGSIG
query Query a BIGSIG on one or more fasta/fastq.gz files
identify Identify reads based on probability
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print versionYou can run the 2 steps to perform read identification like this:
### ref_file_example is path for reference genomes, see below
time bigsig construct -r ref_file_example.txt -b test -k 31 -mv 21 -s 10000000 -n 4 -t 24
time bigsig identify -b test.mxi -q ./test_data/SRR548019.fastq.gz -n output -t 24input format:
### ref_file_example.txt
Salmonella_enterica_salamae_VIII_74-1880 ./refs/74_1880.fasta
Salmonella_enterica_salamae_b_Ar0560 ./refs/Ar0560.fasta
Salmonella_enterica_houtenae_Type ./refs/NCTC10401.fasta
Salmonella_bongori_Type-NCTC12419 ./refs/S_bongoriType_NCTC12419.fastaoutput explain (a PacBio long reads metagenomic dataset): the PREFIX_reads.txt: The first column contains the name of the read, the second column the taxonomic classification (database genome name), the third column the number of minimizers supporting this classification, the fourth column the total number of minimizers used as input for the classification and the fifth column indicates if this classification is rejected or accepted given the false positive probability (default is 0.001). There are 2 cases where a read classification can be rejected: 1. not enough minimzers found in any of the genomes in the database, which mean this the read is from new genus at least, if there are so many such reads, which indicate taxonomic novelty. You should perform reads assembly to obtain population genomes; 2. many minimizers found in multiple genomes, which indicate a very conserved read, read level classification is not reliable in this case.
>m54119U_190516_061409/2/ccs GCA_900546835.1_genomic.fna 980 1099 accept 1
>m54119U_190516_061409/9/ccs GCA_900546835.1_genomic.fna 1015 1342 accept 1
>m54119U_190516_061409/1095/ccs no_significant_hits 0 728 reject 0
>m54119U_190516_061409/73400366/ccs GCA_004562975.1_genomic.fna,GCF_000210075.1_genomic.fna,GCF_000382445.1_genomic.fna,GCA_004555245.1_genomic.fna,GCF_900079775.1_genomic.fna 1351 1358 reject 5The PREFIX_counts.txt summarizes the total counts per taxon. you can use sort and head to get the top hits: sort -grk2 PREFIX_counts.txt | head -10
GCA_900546835.1_genomic.fna 106435
GCA_000437595.1_genomic.fna 83967
GCF_013009555.1_genomic.fna 66368
GCA_025147485.1_genomic.fna 50485
GCF_000012825.1_genomic.fna 40636
GCF_020687245.1_genomic.fna 28853
GCF_000158035.1_genomic.fna 28151
GCA_025146925.1_genomic.fna 28035
GCA_902363145.1_genomic.fna 25749
GCA_900544265.1_genomic.fna 23938
GCF_000233955.1_genomic.fna 23078
GCF_000723465.1_genomic.fna 21830
GCA_025145845.1_genomic.fna 21329
GCF_003461625.1_genomic.fna 17235
GCA_003486385.1_genomic.fna 15961
GCF_003287405.1_genomic.fna 15101
GCA_019414665.1_genomic.fna 14663
reject 14325
GCA_001917135.1_genomic.fna 14149
GCF_023347255.1_genomic.fna 13664
Pre-built binaries will be available on release page binaries for major platforms (no need to install but just download and make it executable). We recommend you use the linux one (gsearch_Linux_x86-64_v0.1.5.zip) for linux system in this release page for convenience (only system libraries are required). For macOS, we recommend the universal binary mac-binaries (gsearch_Darwin_universal_v0.1.5.tar.gz). Or GSearch_pc-windows-msvc_x86-64_v0.1.5.zip for Windows.
### we suggest python 3.8, so if you do not have one, you can create a new one
conda create -n python38 python=3.8
conda activate python38
conda config --add channels bioconda
conda install -c conda-forge -c bioconda gsearch
gsearch -hbrew tap jianshu93/gsearch
brew update
brew install gsearch
gsearch -h
Otherwise it is possible to install/compile by yourself (see install section)
### get the binary for linux (make sure you have recent Linux installed with GCC, e.g., Ubuntu 18.0.4 or above)
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.5/gsearch_Linux_x86-64_v0.1.5.zip --no-check-certificate
unzip gsearch_Linux_x86-64_v0.1.5.zip
cd gsearch_Linux_x86-64_v0.1.5
## get the binary for macOS (universal)
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.5/gsearch_Darwin_universal_v0.1.5.tar.gz --no-check-certificate
tar -xzvf gsearch_Darwin_universal_v0.1.5.tar.gz
## get the binary for Windows, ann subcommand is not available for windows for now
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.1.3-beta/GSearch_pc-windows-msvc_x86-64_v0.1.3.zip
## Note that for MacOS, you need sudo previlege to allow external binary being executed
* **make it excutable (changed it accordingly on macOS)**
chmod a+x ./gsearch
### put it under your system/usr bin directory (/usr/local/bin/ as an example) where it can be called
mv ./gsearch /usr/local/bin/
### check install
gsearch -h
### check install MacOS, you may need to change the system setup to allow external binary to run by type the following first and use your admin password
sudo spctl --master-disable
##We then give here an example of utilization with prebuilt databases.
### download neighbours for each genomes (fna, fasta, faa et.al. are supported) as using pre-built database, probminhash or SetSketch/hll database (prob and hll)
wget http://enve-omics.ce.gatech.edu/data/public_gsearch/GTDBv207_v2023.tar.gz
tar -xzvf ./GTDBv207_v2023.tar.gz
### Densified MinHash database (optdens) for NCBI/RefSeq, go to https://doi.org/10.6084/m9.figshare.24617760.v1
### Download the file in the link by clicking the red download to you machine (file GSearch_GTDB_optdens.tar.gz will be downloaded)
wget http://enve-omics.ce.gatech.edu/data/public_gsearch/GSearch_optdens.tar.gz
mkdir GTDB_optdens
mv GSearch_GTDB_optdens.tar.gz GTDB_optdens/
### get test data, we provide 2 genomes at nt, AA and universal gene level
wget https://github.com/jean-pierreBoth/gsearch/releases/download/v0.0.12/test_data.tar.gz --no-check-certificate
tar xzvf ./test_data.tar.gz
###clone gsearch repo for the scripts
git clone https://github.com/jean-pierreBoth/gsearch.git
### test nt genome database
cd ./GTDB/nucl
##default probminhash database
tar -xzvf k16_s12000_n128_ef1600.prob.tar.gz
# request neighbors for nt genomes (here -n is how many neighbors you want to return for each of your query genome, output will be gsearch.neighbors.txt in the current folder)
gsearch --pio 100 --nbthreads 24 request -b ./k16_s12000_n128_ef1600_canonical -r ../../test_data/query_dir_nt -n 50
## reformat output to have ANI
reformat 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
## SetSketch/hll database
tar -xzvf k16_s4096_n128_ef1600.hll.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k16_s4096_n128_ef1600_canonical_hll -r ../../test_data/query_dir_nt -n 50
## reformat output to have ANI
reformat.sh 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
## Densified MinHash, download first, see above
cd GTDB_optdens
tar -xzvf GSearch_GTDB_optdens.tar.gz
cd GTDB
#nt
tar -xzvf k16_s18000_n128_ef1600_optdens.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k16_s18000_n128_ef1600_optdens -r ../../test_data/query_dir_nt -n 50
## reformat output to have ANI
reformat 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
### or request neighbors for aa genomes (predicted by Prodigal or FragGeneScanRs), probminhash and SetSketch/hll
cd ./GTDB/prot
tar xzvf k7_s12000_n128_ef1600.prob.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k7_s12000_n128_ef1600_gsearch -r ../../test_data/query_dir_aa -n 50
## reformat output to have AAI
reformat 16 1 ./gsearch.neighbors.txt ./clean_AAI.txt
#aa densified MinHash
cd ./GTDB_optdens
cd GTDB
tar -xzvf k7_s12000_n128_ef1600_optdens.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k7_s12000_n128_ef1600_optdens -r ../../test_data/query_dir_aa -n 50
## reformat output to have AAI
reformat 16 1 ./gsearch.neighbors.txt ./clean_ANI.txt
### or request neighbors for aa universal gene (extracted by hmmer according to hmm files from gtdb, we also provide one in release page)
cd ./GTDB/universal
tar xzvf k5_n128_s1800_ef1600_universal_prob.tar.gz
gsearch --pio 100 --nbthreads 24 request -b ./k5_n128_s1800_ef1600_universal_prob -r ../../test_data/query_dir_universal_aa -n 50
###We also provide pre-built database for all RefSeq_NCBI prokaryotic genomes, see below. You can download them if you want to test it. Following similar procedure as above to search those much larger database.
##graph database based on probminhash and setsketc/hll, both nt and aa
wget http://enve-omics.ce.gatech.edu/data/public_gsearch/NCBI_RefSeq_v2023.tar.gz
##nt graph database based on densified MinHash, go to below link to download GSearch_k16_s18000_n128_ef1600_optdens.tar.gz
https://doi.org/10.6084/m9.figshare.24615792.v1
#aa graph database, densified MinHash, go to below link to download GSearch_k7_s12000_n128_ef1600_optdens.tar.gz
https://doi.org/10.6084/m9.figshare.22681939.v1
### Building database. database is huge in size, users are welcome to download gtdb database here: (<https://data.ace.uq.edu.au/public/gtdb/data/releases/release207/207.0/genomic_files_reps/gtdb_genomes_reps_r207.tar.gz>) and here (<https://data.ace.uq.edu.au/public/gtdb/data/releases/release207/207.0/genomic_files_reps/gtdb_proteins_aa_reps_r207.tar.gz>) or go to NCBI/RefSeq to download all available prokaryotic genomes
### build database given genome file directory, .fna/.fasta/.fa/.fna.gz/.fna.xz/.fna.bz2/.fa.gz/.fa.xz/.fa.bz2/.fasta.gz/.fasta.xz/.fasta.bz2 was expected for nt and .faa or .faa.gz/.faa.xz/.faa.bz2 for --aa. Limit for k is 32 (15 not work due to compression), for s is 65535 (u16) and for n is 255 (u8). We recommended optimal densification for its speed and accuracy. --scale_modify_f can be used to adjust number of layers. Hub NSW can be achieved using a small value (e.g.0.25)
gsearch --pio 2000 --nbthreads 24 tohnsw -d db_dir_nt -s 12000 -k 16 --ef 1600 -n 128 --algo optdens --scale_modify_f 0.25
gsearch --pio 2000 --nbthreads 24 tohnsw -d db_dir_aa -s 12000 -k 7 --ef 1600 -n 128 --aa --algo optdens --scale_modify_f 0.25
### When there are new genomes after comparing with the current database (GTDB v207, e.g. ANI < 95% with any genome after searcing, corresponding to >0.875 ProbMinHash distance), those genomes can be added to the database
### old .graph,.data and all .json files will be updated to the new one. Then the new one can be used for requesting as an updated database
gsearch --pio 100 --nbthreads 24 add -b ./k16_s12000_n128_ef1600_canonical -n db_dir_nt (new genomes directory)
### or add at the amino acid level, in the parameters.json file you can check whether it is DNA or AA data via the "data_t" field
cd ./GTDB/prot
gsearch --pio 100 --nbthreads 24 add -b ./k7_s12000_n128_ef1600_gsearch -n db_dir_nt (new genomes directory in AA format predicted by prodigal/FragGeneScanRs)
### visuzlizing from the tohnsw step at amino acid level (AAI distance), output order of genome files are the same with with seqdict.json
cd ./GTDB/prot
gsearch ann -b ./k7_s12000_n128_ef1600_gsearch --stats --embedgsearch.neighbors.txt is the default output file in your current directory.
For each of your genome in the query_dir, there will be requested N nearest genomes found and sorted by distance (smallest to largest) or ANI (largest to smallest). If one genome in the query does not exist in the output file, meaning at this level (nt or aa), there is no such nearest genomes in the database (or distant away from the best hit in the database), you may then go to amino acid level or universal gene level.
-
hnsw_rs relies on the crate simdeez to accelerate distance computation. On intel you can build hnsw_rs with the feature simdeez_f
-
annembed relies on openblas so you must choose between the features "annembed_openblas-static" , "annembed_openblas-system" or "annembed_intel-mkl". You may need to install gcc, gfortran and make. This can be done using the --features option as explained below, or by modifying the features section in Cargo.toml. In that case just fill in the default you want.
-
kmerutils provides a feature "withzmq". This feature can be used to store compressed qualities on a server and run requests. It is not necessary in this crate.
curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh- simple install without annembed feature
cargo install gsearch --no-default-features --features="simdeez_f"- simple installation, with annembed enabled would be with intel-mkl
cargo install gsearch --no-default-features --features="annembed_intel-mkl,simdeez_f"
or with a system installed openblas:
cargo install gsearch --features="annembed_openblas-system,simdeez_f"- On MacOS, which requires dynamic library link (you have to install openblas first):
(note that openblas install lib path is different on M1 MACs).
So you need to run:
brew install openblas
echo 'export LDFLAGS="-L/usr/local/opt/openblas/lib"' >> ~/.bash_profile
echo 'export CPPFLAGS="-I/usr/local/opt/openblas/include"' >> ~/.bash_profile
echo 'export PKG_CONFIG_PATH="/usr/local/opt/openblas/lib/pkgconfig"' >> ~/.bash_profile
source ~/.bash_profile
cargo install gsearch --features="annembed_openblas-system"- direct installation from github:
cargo install gsearch --features="annembed_intel-mkl" --git https://github.com/jean-pierreBoth/gsearch- download and compilation
git clone https://github.com/jean-pierreBoth/gsearch
cd gsearch
#### build
cargo build --release --features="annembed_openblas-static"
cargo build --release --features="annembed_intel-mkl"
###on MacOS, which requires dynamic library link (install openblas first, see above):
cargo build --release --features="annembed_openblas-system"Html documentation can be generated by running (example for someone using the "annembed_openblas-system" feature):
cargo doc --features="annembed_openblas-system" --no-deps --opencargo install --git https://gitlab.com/Jianshu_Zhao/fraggenescanrsSome hints in case of problem (including installing/compiling on ARM64 CPUs) are given here
We provide pre-built genome/proteome database graph file for bacteria/archaea, virus and fungi. Proteome database are based on genes for each genome, predicted by FragGeneScanRs (https://gitlab.com/Jianshu_Zhao/fraggenescanrs) for bacteria/archaea/virus and GeneMark-ES version 2 (http://exon.gatech.edu/GeneMark/license_download.cgi) for fungi.
- Bacteria/archaea genomes are the newest version of GTDB database (v207, 67,503genomes) (https://gtdb.ecogenomic.org), which defines a bacterial speces at 95% ANI. Note that GSearch can also run for even higher resolution species database such as 99% ANI.
- Bacteria/archaea genomes from NCBI/RefSeq until Jan. 2023 (~318,000 genomes)(https://www.ncbi.nlm.nih.gov/refseq/about/prokaryotes/), no clustering at a given ANI threshold.
- Virus data base are based on the JGI IMG/VR database newest version (https://genome.jgi.doe.gov/portal/IMG_VR/IMG_VR.home.html), which also define a virus OTU (vOTU) at 95% ANI.
- Fungi database are based on the entire RefSeq fungal genomes (retrived via the MycoCosm website), we dereplicated and define a fungal speices at 99.5% ANI.
- All four pre-built databases are available here:http://enve-omics.ce.gatech.edu/data/gsearch or at FigShare, see the download links here
Future developments considered are given here
-
Jianshu Zhao, Jean Pierre Both, Luis M Rodriguez-R, Konstantinos T Konstantinidis, GSearch: ultra-fast and scalable genome search by combining K-mer hashing with hierarchical navigable small world graphs, Nucleic Acids Research, 2024;, gkae609, https://doi.org/10.1093/nar/gkae609.
-
Jianshu Zhao, Jean Pierre Both, Konstantinos T Konstantinidis, Approximate nearest neighbor graph provides fast and efficient embedding with applications for large-scale biological data, NAR Genomics and Bioinformatics, Volume 6, Issue 4, December 2024, lqae172, https://doi.org/10.1093/nargab/lqae172